细胞技术有限公司")

依照FDA监管要求开发干细胞药物的中国实践
2020-03-16
您可能在新闻中听说过FDA,或者在其他广告里看到过“FDA认证”的字样,FDA究竟有何厉害之处?为什么“FDA 认证”会被认为是产品品质的最高荣誉和保证?

今天,小编就带您了解一下。
什么是FDA?
FDA是食品药品监督管理局(Food and Drug Administration)的简称,有时也代表美国食品药品监督管理局。FDA由美国国会即联邦政府授权,是专门从事食品与药品管理的最高执法机关,也是一个由医生、律师、微生物学家、化学家和统计学家等专业人士组成的致力于保护、促进和提高国民健康的政府卫生管制的监管机构。其它许多国家都通过寻求和接受 FDA 的帮助来促进并监控其本国产品的安全。
FDA究竟有多权威?
FDA是美国最早的消费者保护机构之一,在美国乃至全球都有极其巨大的影响,有“美国人健康守护神”之称。FDA对其使命和职责的神圣守护与履行,让她成为全球食品药品消费者心中的金刚盾牌。
自1990年以后,美国FDA与ISO组织等国际组织密切合作,不断推动一连串革新措施。尤其在食品、药品领域,FDA认证成为世界食品、药品的最高检测标准。被世界卫生组织认定为最高食品安全标准。只有申报的产品经过对人体使用产品后的143个关键检测点位作监测,对2-3万人持续3-7年的监测,完全通过合格的产品,才会核发FDA认证。
因此,国际很多厂商都以追求获得 FDA 认证作为产品品质的最高荣誉和保证。
美国的新药是如何受FDA监管的?
FDA成立于1906年;之前美国的药品没有任何监管,药品通过广告进行销售;1938年要求对药品证明安全性后,才可以销售;1962年,要求药品不仅有安全性还要证明有效才可销售。FDA有权对生产厂家进行视察、有权对违法者提出起诉。
经过不断的发展,美国FDA已形成了全球最为详细、全面的工作细化布局,共下设12个司局、7个中心。12个司局此处不再赘述,重点说下7个中心(分别是食品安全及应用营养中心、兽药中心、国家毒理研究中心、生物制品评价及研究中心、药草产品中心、药品审评与研究中心、器械及放射卫生中心)中的药品评估和研究中心(CDER)。

美国FDA下设7个中心
CDER是美国FDA最大的审评中心之一,旨在确保处方药和非处方药的安全和有效,是监督《食品、药品和化妆品法》中定义的大多数药品的部门。负责审评新药和非专利药品的申请,通过cGMP规则管理美国的药品生产,确定哪些药物需要医生的处方,同时,该中心还监管电视、广播以及出版物上的药品的广告的真实性,并收集和分析已在市场上有关药品的安全数据,严格监管药品,提供给消费者准确安全的信息。
FDA的审评机制和效率都是值得我们借鉴和学习的,仅在2019年,FDA就批准了48种新药,保持了近几年的发展势头。
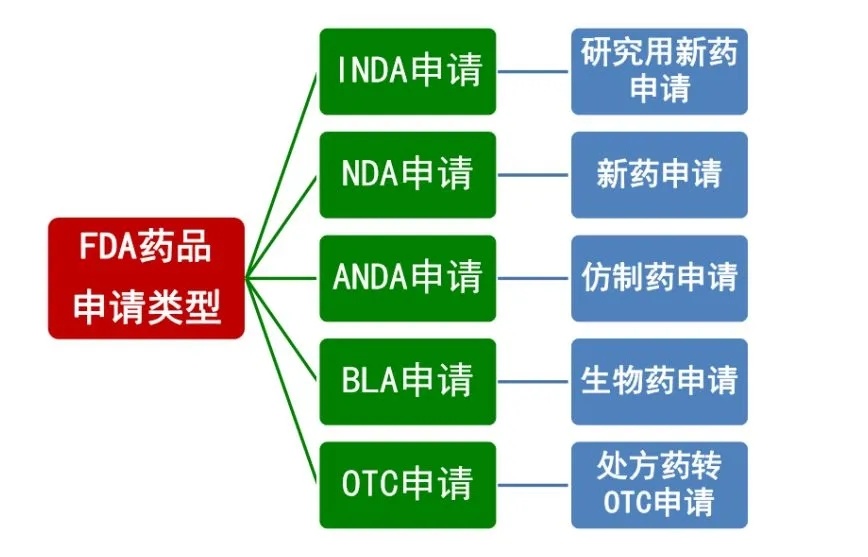
FDA的药品申请类型
《食品药品与化妆品法案》505(i) 规定,对于有资格的专家专为研发所用的药,免除NDA或简化新药上市申报(ANDA)的要求,申请这一豁免被称为新药调查申请(IND)。对于新的IND,要有30天的安全等待期。换句话说,FDA有30天的时间就其安全性做出决定。
IND申请信息须包含(第一阶段最基本的):
1.动物药理学和毒理学研究(安全性需求)
• 保证在人体测试中的安全性
2.生产信息
• 成分、生产商、稳定性、控制措施等
• 保证能生产和供应一致性批次的药品
3.临床方案和研究者信息
IND的各个阶段:
第一阶段: 新药初次用于人类 。该阶段主要评估临床试验所用干细胞药品的安全性,研究药物在人体内的代谢和药学作用,增加剂量引起的不良反应,有效性的早期证据等,受FDA的紧密监视,一般有20-80个病人或正常人参与临床试验。
第二阶段: 受限制和管控的临床研究。第二阶段主要评估临床试验所用药品的初步有效性,一般与安慰剂对比,目的是得到药物有效性的初期数据和短期无严重不良反应的证据。该阶段依然受FDA的紧密监视,受试者通常为几百人。
第三阶段: 扩大的,受管控的和不受管控的实验。此阶段主要评估药品的确切疗效,研究药品的药理作用、代谢、动力学和各种可能性的不良反应,包括联合用药,与已上市同类药物的对比,进行效益/风险评估,目的为得到额外的有效性和安全性信息,为外推到大众,并将此类信息转换为医师标签提供充分基础。通常受试者为几百名到几千名不等。
以上阶段都完成后,就可以进行新药申报了,若能被批准,并且现场检查通过,就能被FDA批准上市了。
九芝堂美科的干细胞符合FDA要求
2020年2月,九芝堂美科干细胞新药临床试验获得批准,这是国家药品监督管理局药物审评中心批准的第一项使用进口干细胞进行的临床试验,第一项使用骨髓来源间充质干细胞进行的临床试验,也是第一项使用干细胞治疗神经系统大适应症的临床试验,对我国干细胞产业的发展具有标志性意义。
本次临床试验所用干细胞产品为美国Stemedica Cell Technologies, Inc.生产的缺血耐受人同种异体骨髓间充质干细胞(ithMSC)。Stemedica公司成立于2005年, 2010年获得了美国药监当局颁发的生产许可证,至今已经在GMP条件下运行了近10年时间,是世界上少数能够在cGMP条件下生产骨髓间充质干细胞和神经干细胞产品的企业,所产干细胞被Life Technology公司(已被赛默飞收购)评为“同类最佳”产品。
Stemedica公司在全程低氧条件下生产的ithMSC产品,其生产工艺和质量体系符合美国cGMP 标准和FDA要求。Stemedica生产的骨髓间充质干细胞产品已在美国获得7项IND,也就是说,这7项临床试验就是在FDA的批准和监管下进行的。
Stemedica及其合作伙伴在全球多个国家开展17项临床试验,涉及卒中、慢性心衰、急性心梗、皮肤光老化、2型糖尿病、骨关节炎、创伤性脑损伤和阿尔茨海默症等多个适应症,其中使用缺血耐受人同种异体骨髓间充质干细胞治疗急性心肌梗死已在哈萨克斯坦完成III期临床试验,获哈萨克斯坦卫生部批准上市。

Stemedica生产的骨髓间充质干细胞和神经干细胞产品全球的研发管线(备注:Stemedica已将心脏领域适应症授权给合作伙伴CardioCell LLC)
九芝堂美科(北京)细胞技术有限公司作为美国Stemedica公司干细胞技术在中国的唯一承接方,通过引进Stemedica全球领先的临床级干细胞制备平台,获得了干细胞生产的规模化、标准化和可追溯性的核心技术,填补国内空白。
美科已在有“中国药谷”之称的北京大兴生物医药基地建成符合中国、美国cGMP标准质量体系的生产平台,可生产符合中、美药品申报要求的商业级干细胞。
接下来,九芝堂美科将与首都医科大学附属北京天坛医院合作开展治疗缺血性脑卒中的临床试验。
上一条:


